
.png)
O desenvolvimento de um novo medicamento além de um processo científico é, sobretudo, um exercício de alocação de capital sob incerteza regulatória.
Entre a descoberta de uma molécula e sua aprovação sanitária, transcorrem em média 10 a 15 anos, com taxas de sucesso extremamente baixas, apenas 1 em cada 10 mil compostos chega ao mercado. Esse funil reflete risco biológico e a complexidade do processo regulatório, que define o ritmo de entrada de inovação no sistema de saúde.
Na prática, cada etapa da fase pré-clínica aos estudos de fase III funciona como um filtro progressivo de risco, com impacto direto em valuation, custo de capital e apetite de investidores.
No Brasil, a ANVISA desempenha um papel central nesse ciclo. Além do órgão técnico, a agência atua como um dos principais determinantes do time-to-market e, consequentemente, da atratividade do país para desenvolvimento clínico e lançamento de novas terapias.
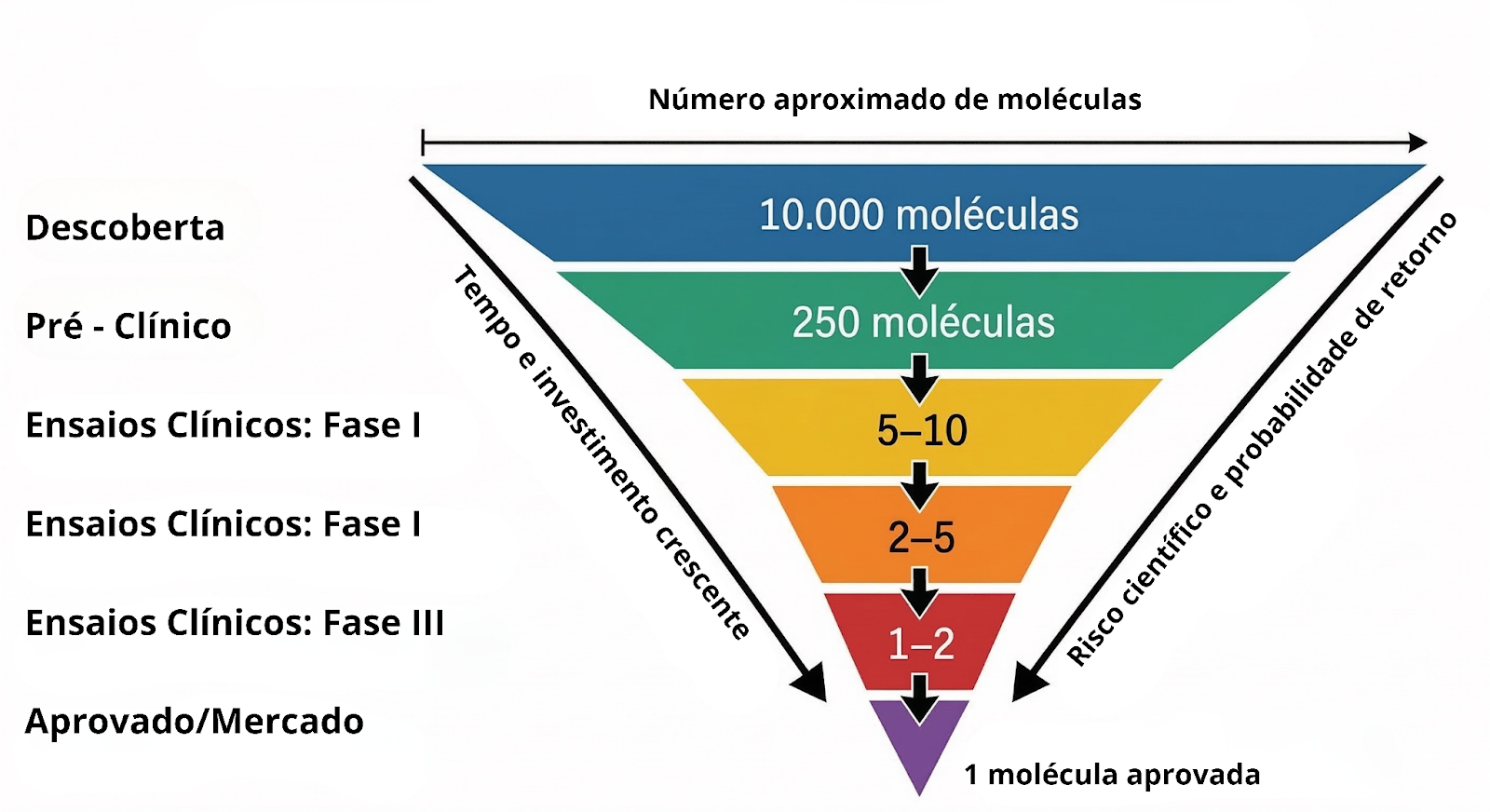
Funil de desenvolvimento de medicamentos
O novo marco regulatório e a tentativa de destravar a inovação
Nos últimos anos, o Brasil passou por uma transformação no seu ambiente regulatório. A combinação da Lei 14.874/2024 com a RDC 945/2024 trouxe mudanças estruturais no processo de aprovação de ensaios clínicos, com maior alinhamento aos padrões internacionais e avanços na segurança jurídica.
Na leitura de Laura Castanheira, que acumulou quase uma década na própria ANVISA e depois liderou a agenda regulatória em grandes farmacêuticas, essas mudanças aproximam o Brasil das melhores práticas globais e corrigem distorções históricas do sistema.
“A RDC 945/2024 representa um avanço significativo para o Brasil aumentar expressivamente sua competitividade global na área de ensaios clínicos.”
Um dos principais gargalos, o tempo de aprovação, começa a ser endereçado. A expectativa é de redução relevante nos prazos para início dos estudos, algo que, segundo Laura, historicamente afastava o país de oportunidades relevantes.
Ela destaca que o aprimoramento do processo regulatório tende a reduzir perdas de estudos internacionais, problema recorrente no passado, quando atrasos inviabilizavam a inclusão do Brasil em protocolos globais.
Hoje, com prazos mais previsíveis e processos mais estruturados, o país passa a disputar de forma mais competitiva o fluxo global de pesquisa clínica.
Velocidade vs. segurança
A modernização regulatória ocorre em paralelo a uma pressão crescente por acesso mais rápido a novas terapias, especialmente em áreas como obesidade, oncologia e doenças raras. Esse movimento exige uma mudança de abordagem: acelerar sem abrir mão do rigor técnico.
Nesse contexto, a ANVISA tem adotado instrumentos que aumentam eficiência sem comprometer a qualidade da análise. Entre eles, ganham destaque as reuniões de pré-submissão com empresas e, principalmente, o uso do chamado reliance regulatório.
Como explica Laura, esse mecanismo permite que o Brasil considere avaliações de agências internacionais de referência, evitando retrabalho e reduzindo o tempo de decisão.
“Esse procedimento permite tornar a análise mais ágil, sem abrir mão da qualidade e rigor técnico.”
Na prática, a regulação passa a operar de forma mais integrada globalmente, um movimento essencial diante da crescente complexidade dos novos medicamentos.
Quando a regulação não acompanha a demanda: o caso da retatrutida
Se por um lado a regulação avança, por outro a velocidade da demanda, impulsionada por redes sociais, tendências globais e pressão de pacientes, começa a ultrapassar o ritmo institucional. O caso da retatrutida ilustra esse desalinhamento.
Ainda em fase de estudos clínicos e sem aprovação sanitária, a molécula já vem sendo comercializada de forma irregular em mercados paralelos, incluindo países vizinhos como o Paraguai. O fenômeno evidencia um novo tipo de risco: o acesso antecipado, e não autorizado, a terapias experimentais.
Do ponto de vista regulatório, no entanto, as regras são claras. Como reforça Laura, a manipulação de substâncias no Brasil exige que a molécula já tenha passado por avaliação completa de qualidade, segurança e eficácia.
“Uma condição obrigatória é que sejam moléculas devidamente registradas na Anvisa.”
Além da base normativa, o sistema de vigilância sanitária atua de forma ativa na fiscalização dessas práticas. Em casos mais sensíveis, a agência tem ampliado o nível de exigência regulatória, especialmente para classes terapêuticas sob maior pressão de demanda, como os agonistas de GLP-1.
Laura aponta que, nesses casos, a ANVISA tem adotado medidas adicionais, como reforço na exigência de prescrição médica e maior controle sobre a qualidade dos insumos importados utilizados na manipulação.
O que está em jogo: competitividade, inovação e risco sistêmico
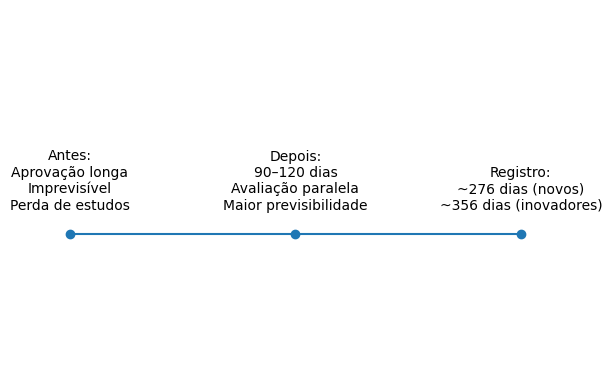
Timeline regulatório – Ensaios Clínicos (Brasil)
O Brasil entra, assim, em um momento de inflexão regulatória. Os avanços recentes, como maior alinhamento internacional, redução de prazos e aumento de previsibilidade, colocam o país em uma posição mais competitiva para atrair ensaios clínicos globais e participar de forma mais relevante na cadeia de inovação farmacêutica.
Ao mesmo tempo, esse novo contexto traz desafios igualmente relevantes. A pressão por acesso acelerado a novas terapias, combinada com a disseminação de informações em escala digital, tem contribuído para o surgimento de mercados paralelos e ampliado o risco sanitário, especialmente em áreas de alta demanda como obesidade e oncologia.
Nesse cenário, a agenda regulatória passa a ocupar um papel estratégico. O desafio não é mais simplesmente controlar, mas coordenar, equilibrando a velocidade da inovação com a necessidade de garantir segurança, qualidade e eficácia. Em última instância, a forma como esse equilíbrio será conduzido tende a definir o acesso da população a novos tratamentos e o posicionamento do Brasil na dinâmica global da indústria farmacêutica.
A Green Rock é uma gestora de investimentos independente, focada em negócios de Venture Capital e Private Equity de alto potencial no setor da saúde do Brasil e toda a América Latina.
.png)
